CRANIOFACIAL DISEASES

Craniofacial diseases and abnormalities are those conditions which involve the skull and upper face. The speciality is multi disciplinary where several specialists from different disciplines manage these conditions.
CLASSIFICATION
Craniofacial diseases are classified into the following categories depending whether the major abnormality involves premature fusion of the craniofacial sutures or due to deficiencies and defects in the craniofacial bony structure.
CRANIOSYNOSTOSES
In this condition there is premature fusion of the cranial sutures resulting in disproportionate growth of the cranial bones and as a sequence the growth of the facial bones. When a suture is fused there is no growth in a plain perpendicular to the line of the suture.
Skull has two different components, membranous part which comprises the vault, and chondrocranium which comprises the base of the skull. After a certain age no regrowth occurs in skull defects in the membranous skull contrary to the chondrocranium.
It is particularly important to indicate that brain growth induces the osteogenesis of the skull and this occurs in a homogeneous and symmetrical manner. The skull grows rapidly from birth to 7th year but the greater part of the increase of its cranial part occurs is during the first year owing to the rapidity of the growth in that period of the brain The brain reaches its maximum growth at about the age of two years.
There are six fontanelles at the edges of the parietal bones. Two anterolateral and posterolateral as well as anterior and posterior fontanelle. The posterior and anterolateral fontanelles are obliterated within two to three months after birth. The posterolateral fontanelle is usually closed about the end of the first year and the anterior fontanelle about the middle of the second year..
Obliteration of the sutures of the vault of the skull takes place as age advances. It commences between the ages of 30 and 40 on the inner surface of the skull and about 10 years later on the outer surface of the skull. Obliteration usually occurs first in the lower part of the coronal suture, next in the posterior part of the sagittal suture and then in the lambdoid suture.
The major skull sutures are:
1 Metopic or frontal suture which separates the frontal bone into two halves. Union starts in the first year and is completed by the 8th year.
2 The sagittal suture, this separates the two parietal bones and extends from the anterior fontanelle to the posterior fontanelle or from bregma to lambda.
3 The coronal suture separates the frontal bone from the parietal bones.
4 The lambdoid suture, this separates the posterior edge of the parietal bone from the occipital bone.
5 Squamosal suture which is the superior border of the squamous part of the temporal bone. Anteriorly it articulates with the greater wing of sphenoid, superiorly it articulates with the parietal bone and posteriorly and inferiorly it articulates with the occipital bone.
FUSION
The time of closure of sutures varies widely. The metopic suture closes during the first year of life. The squamosal, occipitomastoid and sphenotemporal sutures may still be partly open until late in life. At birth the body of the sphenoid is separated from the basioccipital by sphenooccipital synchondrosis at which the main growth of the base of skull occurs, as well as the sutures which separate the sphenoid and the ethmoid and the frontal parts of the skull base. There are two types of joints that separates the skull bones, syndesmosis where fibrous tissue separates the bones and synchondrosis where cartilage separates the bones. These are joints but without a joint cavity. In the base of the skull we have synchondrosis while in the vault we have syndesmosis.
A popular theory about the cause of skull deformity and premature suture fusion is that the pathology starts in the sutures of the skull base.
PATHOGENESIS OF CRANIOSYNOSTOSIS
Premature fusion of sutures is bony closure of the affected suture. Other than the metopic suture all sutures should remain open during infancy and childhood. The aetiology behind craniosynostosis is obscure in majority of cases. Genetic disorder is the cause behind some of the complex craniosynostoses. Even in these cases many of them are spontaneous mutation rather than congenital..
Rarely ,metabolic disorders can cause craniosynostosis as in hyperthyroidism.
Craniosynostosis is classified into two main categories:
1 Simple or unisutural craniosynostosis
2 Complex or syndromal craniosynostosis.
INCIDENCE
It is very difficult to assess the incidence of craniosynostosis in the general population as there are many unreported cases. In South Australia Professor Donald Simpson et al reported a rate of 1 per 400,000 births but the figure varied over the years as awareness of the condition increases in the community.

The main presenting features of craniosynostosis are as follows:
1 Appearance, the child is born with obvious deformity depending on the type of craniosynostosis.
2 Raised intracranial pressure, in some cases of craniosynostosis premature suture fusion can lead to reduced skull volume and signs of raised intracranial pressure, this could be in the form of headaches, visual failure and mental retardation.
3 Exophthalmos, this mainly occurs in cases of syndromal synostoses.
4 Orbital hypertelorism or hypotelorism.
5 Orbital dystopia
6 Airway obstruction due to mid face hypoplasia and compromise of the post nasal spaces.
As mentioned above, craniosynostosis is classified into two main categories:
In Simple or unisutural craniosynostosis, there is premature fusion of a single suture. These cases are usually not familial neither do they have genetic basis.
In complex syndromal multisutural craniosynostosis, there are more than one suture involved. They have genetic basis and often familial. Although many of these cases do not present with previous family history. These cases also are associated with other systemic abnormalities.
SIMPLE CRANIOSYNOSTOSIS
In these cases only a single suture is involved and the deformity depends on the suture involved. The main presentation is skull deformity, raised intracranial pressure is not a feature of these conditions. The deformity is usually noticed at birth, although sometimes it is not obvious because of the head swelling associated with some difficult deliveries.
Scaphocephaly:
Here we have premature fusion of the sagittal suture, as mentioned before there is no growth in a plain perpendicular to the line of the suture and skull growth is mainly in anteroposterior direction. On examination there is an overhanging protuberant forehead with associated protuberance of the occipital bone. There are no systemic abnormalities associated with the deformity. The deformity is noticed at birth and medical advice is usually sought soon after.
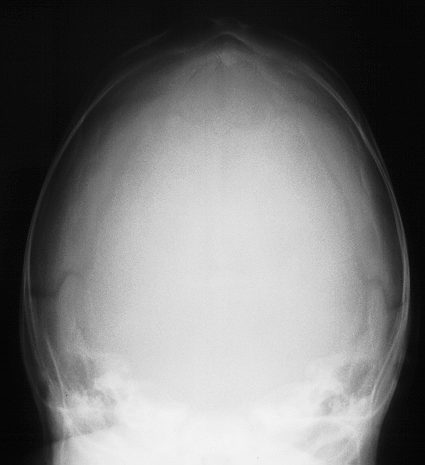
Scaphocephaly fused sagittal suture
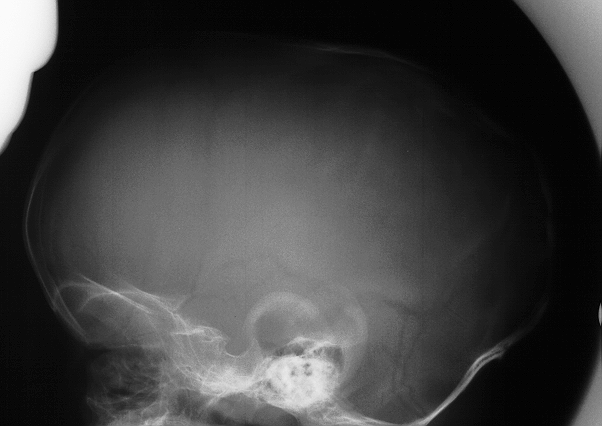
Scaphocephaly

Fused Sagittal suture
MANAGEMENT
This is usually surgical. Up to the age of three months sagittal craniectomy with extension of the craniectomy into the coronal and lambdoid suture leads to significant improvement in the head shape in the ensuing months.
In this procedure a sagittal scalp incision extending from the anterior to the posterior fontanelle is made. A single piece of the skull is removed measuring 5 cms in width and extends from the anterior fontanelle to the posterior fontanelle. Following that the craniectomy is extended both into the coronal and the lambdoid sutures.
Although the craniectomy is extensive reformation of bone and the closure of the skull defect occurs within 4 to 6 months after the procedure. In some cases this procedure does not result in significant improvement of head shape, especially if the procedure is done later than 4 to 5 months of age, also in untreated cases which present later in childhood. In both of these categories complete vault reshaping is necessary. This definitive treatment results in a near normal head shape.
The procedure of vault reshaping is a major procedure with significant risks and should only be done in well equipped Craniofacial Units.


vault reshaping and end result
Trigonocephaly
The metopic suture is usually open at birth and fusion occurs in the first 12 months of life. Premature fusion of this suture results in trigonocephaly. In these cases the forehead is narrow with a prominent mid-frontal ridge. Associated with this there is abnormality of the supraorbital margins and at sometimes hypotelorism. These changes result in a definite recognisable deformity of the forehead and eyes.

Trigonocephaly Fused Metopic suture
With this deformity there are no associated systemic changes nor any risks of raised intracranial pressure.
This deformity has no genetic basis nor any familial history.
MANAGEMENT
The definitive treatment is surgical correction of the deformity. This is recommended to be done in the first six months of life and involves bilateral lateral canthal advance and excision of the abnormal metopic suture.
The procedure is done through a bicoronal scalp incision. Bifrontal craniotomy is performed including the fused metopic suture. The metopic suture is excised and the frontal bone is reshaped. Following that the supraorbital margin is removed and is corrected from an angled position to a flat symmetrical position. The frontal bone is then replaced and fixed to the reformed supraorbital margin to give a symmetrical flat forehead.

Trigonocephaly postop correction
BICORONAL SYNOSTOSIS OR TURRICEPHALY
In this condition there is bilateral fusion of the coronal sutures resulting in abnormally tall and broad head with a narrow anteroposterior diameter. The deformity is characteristic and is not associated with other systemic changes. The condition usually has no genetic basis although familial cases have been reported in the literature. Bony ridges usually could be felt along the line of the fused suture.

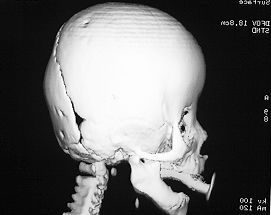
Brachycephaly
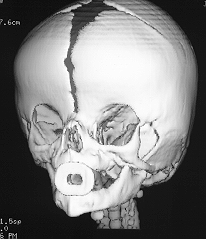
fused coronal sutures

post fronto-orbital advance
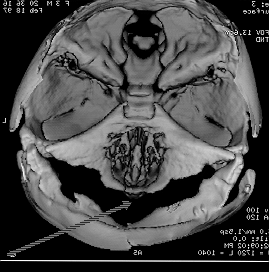
shallow ant cranial fossa and post fossa
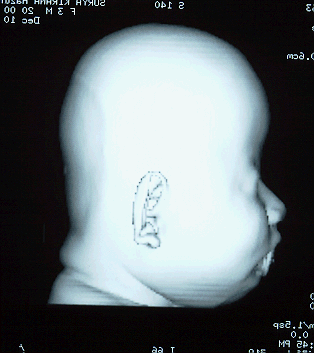
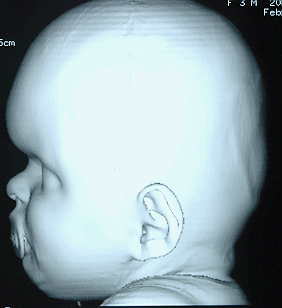
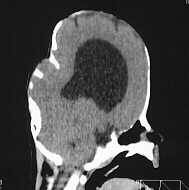
Turricephaly in bicoronal synostosis preop and postop
MANAGEMENT
Definitive management is surgical. The operation is performed by a bicoronal scalp incision to expose the fused coronal suture. The exposure should extend into the temporal fossa. Some surgeons are satisfied by simply removing the coronal sutures and extensive craniectomy across the squamosal suture towards the base of the skull. However it is more acceptable that in addition to the craniectomy frontal orbital advance is performed. Bifrontal craniotomy is done and the frontal orbital bar is excised and fixed in an advanced position. This would improve the overall head shape and increase the length of the skull.
PLAGIOCEPHALY
In plagiocephaly there are associated cranial and facial deformities.
Plagiocephaly is classified into:
1 Frontal plagiocephaly caused by unilateral coronal synostosis.
2 Occipital plagiocephaly caused by unilateral synostosis of the lambdoid suture.
3 Parallelism: in these cases a distinct deformity is noted where the frontal region is depressed on one side with the occipital deformity on the opposite side. This is a common deformity seen in early infancy.
FRONTAL PLAGIOCEPHALY
This is caused by premature fusion of the coronal suture. In this case we have a characteristic deformity where the frontal bone is depressed on the affected side with an apparent bulge of the normal side. The supraorbital margin is also deviated backward and upward. These conditions are associated with lower face twist. The pathological changes extends from the coronal suture into the base of the skull which accounts for the lower facial deformity. This disorder is not congenital , has not a genetic basis and is not usually associated with raised intracranial pressure. The ipsilateral ear is usually situated more anterior and inferior than usual.


Plagiocephaly and fused Coronal sutures
A characteristic feature of fronto plagiocephaly is the radiological appearance on plain skull x-ray where the supraorbital margin sweeps backward and upward, in what is described as Harlequin Deformity. The sphenoidal margin is also seen to sweep upward parallel to the supraorbital margin. The ipsilateral coronal suture is fused and examination would reveal a palpable ridge overlying that suture.
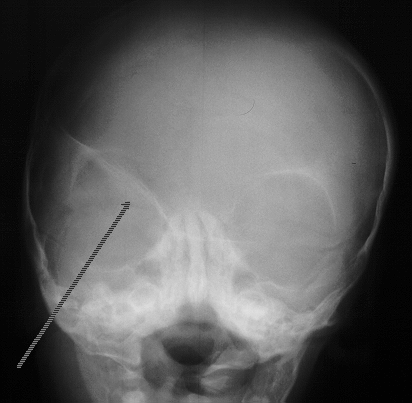
 Small acf
Small acf
Orbital deformity in Frontal plagiocephaly
MANAGEMENT
Definitive management is surgical where the affected coronal suture is excised, it is important to extend the excision down to the base of the skull .In addition to suture excision , fronto-orbital advance on the ipsilateral side is done to bring the forehead in a position symmetrical to the unaffected side. The operation is combined with osteotomy along the fronto-sphenoidal suture in the anterior cranial fossa.
OCCIPITAL PLAGIOCEPHALY
In this condition there is premature fusion of the lambdoid suture. Isolated occipital plagiocephaly is uncommon and is not associated with other deformities as in frontol plagiocephaly. Most of occipital deformity are seen as part of parallelism which will be described later on. . Examination would reveal a flattening of one occiput with an apparent bulging of the normal side.
X-rays of the skull would reveal unilateral premature fusion of the lambdoid suture.
MANAGEMENT
Surgery is the definitive treatment, where lambdoid craniectomy is done . However in our experience we found that the best surgical outcome results from reshaping of the occipital bone.
PARALLELISM
This is a condition which is frequently seen in infants where there is a flattening of one frontal bone and deformity of the contralateral occipital lobe. This is a common condition and is not usually associated with sutural abnormality. We believe that this is mostly related to torticollis or postural in origin. Examination would reveal flattening of one frontal bone with flattening of the contralateral occipital bone. There is also abnormality in the position of the ear. The ear on the frontal deformity side would be placed more posteriorly. There is no other associated deformity and there is no facial deformity.
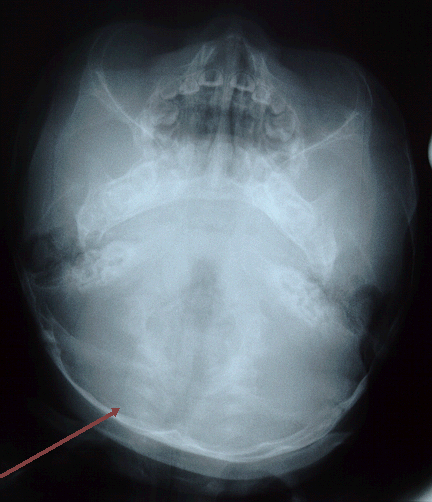
Occipital deformity

normal lambdoid sutures
MANAGEMENT
These cases are managed conservatively in early age, by treating the torticollis and by head positioning. The parents are encouraged to let the child to lie on the prominent side of the head. In most cases improvement occurs with conservative management . However if the deformity persisted or the deformity is severe or was not treated early, surgical correction could be performed if requested by the family.

Deformity improved ,3 weeks after conservartive management

CRANIOFACIAL SYNDROMES
Craniofacial syndrome includes those conditions in which there is abnormal growth of the skull associated with abnormalities of facial growth.
The craniofacial syndromes are usually familial and in many cases abnormal gene were isolated. Several syndromes have been described, only the common ones will be discussed.
CROUZON SYNDROME
This was first described by Crouzon in 1912. He described three major abnormalities, skull deformity, facial deformity and exophthalmos.
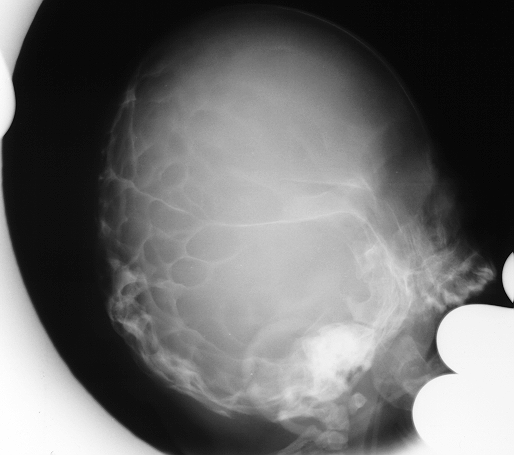
Crouzon disease at birth
The skull typically brachycephalic, there is a prominent nose and protruding jaw. Maxillary hypoplasia is also a prominent feature. Exophthalmos is often severe and pose definite risk to vision.
Crouzon is the commonest craniofacial syndrome.
Skull Deformities
Skull deformities vary greatly but on the whole the skull is brachycephalic .Turricephaly or oxycephaly can occur. There is diffuse premature effusion of the skull sutures including the basal sutures.

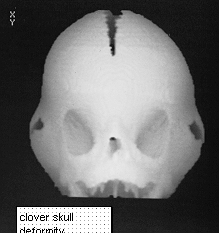
Turricephaly in Crouzon
FACIAL DEFORMITIES
Typically the patient has a peaked pointed nose, exophthalmos of variable degrees and maxillary hypoplasia. The jaw is prominent with malocclusion.
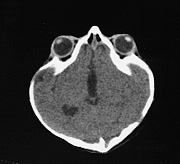
The exophthalmos is due to shallow small orbits which could result in corneal ulceration and conjunctival infection. In some cases the exophthalmos is so severe that eyes are dislocated outside the orbit with imminent loss of vision. As a result of maxillary hyperplasia there is usually compromise of the nasal airways and respiratory difficulties. In addition to sutural premature fusions x-ray often shows a copper beaten appearance which could be an indicator of raised intracranial pressure. Raised intracranial pressure is not an unusual association with this syndrome. Hydrocephalus is also present in about 20% of cases.
severe exophthalmos ,very shallow orbits
Inheritance is of mendelian dominant of high penetrance. However sporadic cases can also occur.
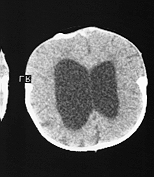
Hydrocephalus in Crouzon
MANAGEMENT
Treatment of Crouzon Syndrome is complex and involves several aspects.
1 Management of skull deformity. This is achieved by frontal orbital advance.
2 Management of exophthalmos and shallow orbits. Frontal orbital advance increases the volume of the orbits so the eye globes are better accommodated within the orbits.
3 Management of maxillary hypoplasia and obstructive airways. The definitive treatment for this condition is mid facial advance ,however this can only be done at later age, when the child reaches 8 or 9 years old.
4 Management of raised intracranial pressure and hydrocephalus. Frontal orbital advance increases the skull volume and helps to relieve raised intracranial pressure. If this was not achieved by this bilateral decompressive craniotomies should be done. If hydrocephalus is present shunt should be inserted.
5 Management of speech.
6 Orthodontic management.
As seen from the above ,care of craniofacial syndromes is a complex and very specialised treatment which spans over several years.
APERT SYNDROME
This syndrome presents with very high brachycephalic head and severe syndactyl affecting all limbs. The syndactyl involves bony fusion. Apert is the second most common syndrome after Crouzon.

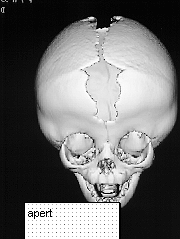
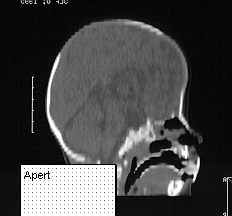
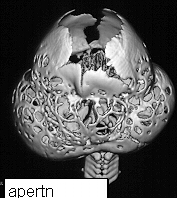
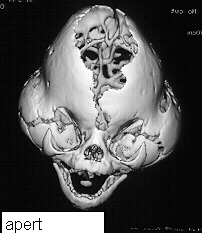
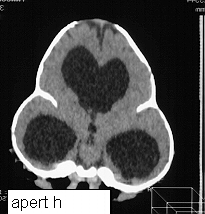
On presentation the head is brachycephalic and turricephalic with widely opened anterior fontanelle and metopic suture. The coronal sutures are well fused at birth . Premature fusion of squamosal and sagittal sutures can also be seen. Typically there is a high forehead and flattened mid face. The maxilla is hypoblastic but exophthalmos is not as severe as in Crouzon. Associated clefts of soft palate and uvula are often seen. Varying degree of mental retardation is often associated with this syndrome as well as hydrocephalus.
Hands and feet shows bony fusion.

Apert is an autosomal dominant trait but most cases are sporadic.
MANAGEMENT
This is essentially to improve headshape by frontal orbital advance and treatment of hydrocephalus. Surgery to the hands is also an essential part of management.

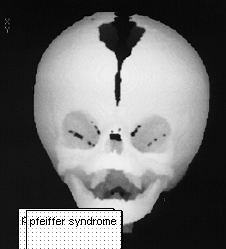
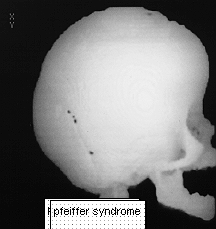
PFEIFFER SYNDROME
This was first described in 1964.THis syndrome has autosomal dominance with high penetrance but sporadic cases do occur.
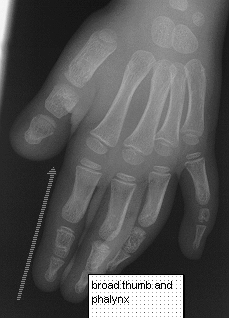
The most common deformity is turricephaly associated with premature fusion of the coronal sutures as well as other sutures. Hypertelorism is common with mild exophthalmos. Occasionally hypoplasia of the maxilla is seen. Characteristically in this syndrome the thumbs are short and broad as well as the great toes with square first phalanges.
Management would consist of correction of head asymmetry by frontal orbital advance.

There are syndromes that are less common like Saethre-Shotzen Syndrome, Carpenter Syndrome and Cohen Syndrome.
back to index